
Thalassemia
Definition
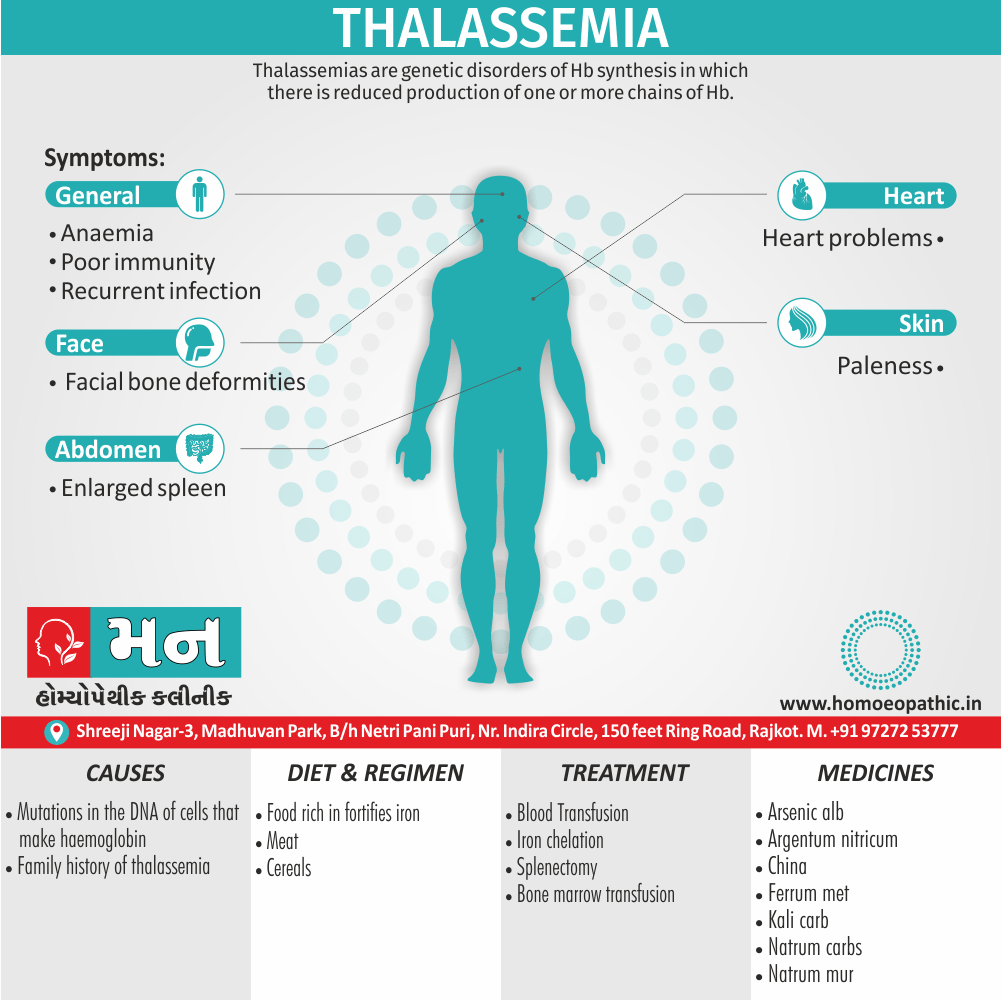
Thalassemia are genetic disorders of Hb synthesis in which there is reduced production of one or more chains of Hb.
It is a genetic blood disorder characterized by reduced or absent production of hemoglobin, the protein in red blood cells responsible for carrying oxygen. It doesn’t have exact synonyms, but Thalassemia known by other names, particularly those that specify the type or severity:
- Cooley’s anemia: This refers to the most severe form of beta-thalassemia, also known as beta-thalassemia major.
- Mediterranean anemia: This is a general term for thalassemia, reflecting its prevalence in populations around the Mediterranean Sea.
- Alpha-thalassemia: This type affects the production of alpha-globin chains in hemoglobin.
- Beta-thalassemia: This type affects the production of beta-globin chains in hemoglobin.
The severity of it varies depending on the specific genetic mutation and the number of affected genes. Milder forms may cause mild anemia, while severe cases can lead to significant health problems, including growth delays, bone deformities, and organ damage.
Overview
Epidemiology
Causes
Types
Risk Factors
Pathogenesis
Pathophysiology
Clinical Features
Sign & Symptoms
Clinical Examination
Diagnosis
Differential Diagnosis
Complications
Investigations
Treatment
Prevention
Homeopathic Treatment
Diet & Regimen
Do’s and Don'ts
Terminology
References
Also Search As
Overview
Overview:
This results in a relative excess production of either α chains or β chains, which without their partner chains are unstable and precipitate in RBCs or their precursors.
The inclusion bodies produced by this process increase the rigidity of RBCs and result in their destruction, either in the marrow or the circulation or both.
Hence the anemia of thalassemia results from ineffective erythropoiesis due to intramedullary RBC destruction, and a shortened RBC survival caused by hemolysis.[2]
Epidemiology
Epidemiology:
The prevalence of β-thalassemia trait in India is estimated to be around 3-4% on average, but higher frequencies have been observed in certain communities, including Sindhis, Punjabis, Gujaratis, Bengalis, and others ("Prevalence and Management of β-Thalassemia in India", Tandfonline, 2021).
Regional variations in prevalence have also been noted. Studies indicate that Northern and Western Indian states tend to have a higher thalassemia burden, while in Eastern India, tribal populations show a higher prevalence compared to non-tribal populations ("Prevalence and Management of β-Thalassemia in India", Tandfonline, 2021).
Specific prevalence data can vary depending on the study and region. For instance, the prevalence of β-thalassemia trait in Central India has been reported to range between 1.4 and 3.4%, while in South India, it ranges from 8.50 to 37.90% ("Prevalence and Management of β-Thalassemia in India", Tandfonline, 2021).[3]
Causes
Causes
It is caused by mutations in the DNA of cells that make hemoglobin — the substance in red blood cells that carries oxygen throughout your body. The mutations associated with thalassemia are passed from parents to children.
Hemoglobin molecules are made of chains called alpha and beta chains that can be affected by mutations. In thalassemia, the production of either the alpha or beta chains are reduced, resulting in either alpha-thalassemia or beta-thalassemia.
In alpha-thalassemia, the severity of thalassemia you have depends on the number of gene mutations you inherit from your parents. The more mutated genes, the more severe your thalassemia.
In beta-thalassemia, the severity of thalassemia you have depends on which part of the hemoglobin molecule is affected.
Alpha-thalassemia
Four genes are involved in making the alpha hemoglobin chain. You get two from each of your parents. If you inherit i.e.:
- One mutated gene-you’ll have no signs or symptoms of thalassemia. But you are a carrier of the disease and can pass it on to your children.
- Two mutated genes-your thalassemia signs and symptoms will be mild. This condition might be called alpha-thalassemia trait.
- Three mutated genes- your signs and symptoms will be moderate to severe.
Inheriting four mutated genes is rare and usually results in stillbirth. Babies born with this condition often die shortly after birth or require lifelong transfusion therapy. In rare cases, a child born with this condition can be treated with transfusions and a stem cell transplant.
Beta-thalassemia
Two genes are involved in making the beta hemoglobin chain. You get one from each of your parents. If you inherit i.e.:
- One mutated gene-you’ll have mild signs and symptoms. This condition is called thalassemia minor or beta-thalassemia.
- Two mutated genes-your signs and symptoms will be moderate to severe. This condition is called thalassemia major, or Cooley anemia.
Babies born with two defective beta hemoglobin genes usually are healthy at birth but develop signs and symptoms within the first two years of life. A milder form, called thalassemia intermedia, also can result from two mutated genes.[2]
Types
Classification:
Thalassemia’s are classified, according to the particular globin chain that ineffectively produce. In α thalassemia there reduce rate of chains synthesis. β thalassemia’s associate with a deficiency of β chains.
In β thalassemia’s, a chain synthesis continues beyond the neonatal period, and most patients have an increased proportion of HbF (a2g2).
α thalassemia, the imbalance produces an excess of β chains, which form β tetramers (HbH), in adult life, and an excess of γ chain, which form γ tetramers (HbBart’s) in infancy.
Molecular Basis of the thalassemia i.e.
- Molecular biology has provided an understanding of the nature of the defects in thalassemia syndromes. There are two a genes present on each chromosome 16 – a1 and a2.
- The β genes are located on chromosome 11 alongside the g and d genes.
- A large number of molecular defects are associated with a thalassemia phenotype, most of the defects in β thalassemia are small, often single base- pair changes.
- There is marked phenotype variation in this condition because of heterogeneity at the molecular level. Some patients have mutations that result in failure to produce any globin chain whereas other mutations allow globin chain production at a reduced rate.
- In α thalassemia, gene deletions are the most common molecular defects, but, because any individual inherits four genes, there are broadly four phenotypes corresponding to deletion of one, two, three or four genes.
- β – thalassemia Occur predominantly in Mediterranean region and Middle and Far East. Clinically, β thalassemia is a condition of variable severity, ranging from the most severe thalassemia, to the mild heterozygous forms in which the patients have mild microcytic hypochromic anemia.
β- Thalassemia:
β-thalassemia Major – (Homozygous β-thalassemia)
Special tests for diagnosis i.e.
- HbF levels are high since γ-chain synthesis continues in absence of β-cell formation.
- Hb electrophoresis – demonstrates bands of both HbA and HbF in β-thalassemia.
- Globin chain synthesis show lack of synthesis of β-chains hence a: b globin chain synthesis ratio is altered (normal 1:1).
- DNA analysis is useful in assessing the severity of the disease and diagnosis.
- Bio-magnetic liver spectrometry evaluates hemosiderosis of the liver.
Thalassemia Intermedia:
- Patients who inherit mutations which lead to only mild reduction in β chain synthesis have a much less severe disorder.
- They are usually not transfusion dependent, though splenomegaly and hypersplenism may warrant splenectomy.
Heterozygous β-thalassemia (β-thalassemia trait):
- Those affected are usually asymptomatic and diagnosis is made on discovery of hypochromic anemia in absence of iron deficiency or other causes.
- Mild to moderate splenomegaly in about one-third. HbA2 elevated.[2]
Risk Factors
Risk factor:
Factors that increase your risk of it include i.e.:
- Family history- It is passed from parents to children through mutated hemoglobin genes.
- Certain ancestry- It occurs most often in African Americans and in people of Mediterranean and Southeast Asian descent.[2]
Pathogenesis
Pathogenesis:
"They are caused by inherited mutations that decrease or abolish the synthesis of one or more of the globin chains that compose adult hemoglobin (Hb A; α2β2), resulting in a hypochromic, microcytic anemia. The decreased synthesis of one globin chain leads to a relative excess of the other chain(s), and the accumulation of these unpaired chains within the developing red cell has deleterious effects on red cell survival. In β-thalassemia, there is a relative excess of α-globin chains, which form insoluble aggregates that precipitate within the red cell, causing membrane damage and ineffective erythropoiesis (the premature death of red cells in the bone marrow). In α-thalassemia, there is a relative excess of β-globin chains (and in the fetus and newborn, also γ-globin chains). These unpaired chains form soluble but unstable tetramers (β4 or γ4), which have abnormal oxygen-binding properties."[4]
Pathophysiology
Pathophysiology:
- Accumulation of free α-chains in norm oblasts which fail to mature and die in the marrow (apoptosis) which results in ineffective haemopoiesis.
- Extravascular hemolysis – Red cells formed from abnormal norm oblasts are destroyed in the spleen causing hemolytic anemia.
- Changes in bone marrow and bones – Development of anemia due to short red cell survival, stimulates EPO production which acts on bone marrow leading to erythroid hyperplasia of bone marrow causing expansion of medullary cavities of bones, widening of both outer and inner tables of the skull and of long bones.
- Extramedullary haemopoiesis causes hepatosplenomegaly.
- Iron overload is due to hemolysis of RBCs, increased absorption of iron from GI tract, repeated blood transfusions. Iron gets deposited in endocrines, liver, heart, bone marrow, spleen, pituitary and islets of Langerhans.[2]
Clinical Features
Clinical Features:
- Anemia: The hallmark of thalassemia is anemia, which results from ineffective erythropoiesis and shortened red blood cell lifespan. The severity of anemia ranges from mild to profound, depending on the specific thalassemia subtype.
- Splenomegaly: Enlarged spleen is a common finding in thalassemia due to increased workload in removing damaged red blood cells.
- Hepatomegaly: Enlarged liver can occur due to extramedullary hematopoiesis and iron overload.
- Bone Changes: Expansion of bone marrow in response to increased erythropoietic demand can lead to characteristic skeletal deformities, particularly in the skull and facial bones ("chipmunk facies").
- Growth Retardation: Chronic anemia and hypoxia can impair growth and development, particularly in children with severe thalassemia.
- Iron Overload: Repeated blood transfusions or increased iron absorption can result in iron overload, which can damage vital organs such as the heart, liver, and endocrine glands.[5]
Sign & Symptoms
Signs and Symptoms :
- Anemia: Reduced red blood cell count leading to fatigue, weakness, and paleness.
- Jaundice: Yellowing of the skin and eyes due to the breakdown of red blood cells.
- Splenomegaly: Enlarged spleen, which may cause abdominal discomfort.
- Hepatomegaly: Enlarged liver.
- Bone deformities: In severe cases, thalassemia can cause changes in the bone structure, particularly in the face and skull.
- Growth retardation: Delayed growth and development in children.
- Heart problems: In severe cases, thalassemia can lead to heart failure.[6]
Clinical Examination
Clinical Examination:
- General Appearance: Patients with thalassemia may have a pale complexion, jaundice (yellowing of the skin and eyes), and splenomegaly (enlarged spleen).
- Growth and Development: Children with thalassemia may have delayed growth and development.
- Cardiovascular System: Patients with thalassemia may have tachycardia (fast heart rate) and heart murmurs.
- Respiratory System: Patients with thalassemia may have shortness of breath and decreased exercise tolerance.
- Musculoskeletal System: Patients with thalassemia may have bone pain and fractures.
- Neurological System: Patients with thalassemia may have headaches and dizziness.[6]
Diagnosis
Diagnosis
- Nessoft test – is naked eye red cell osmotic fragility test. In case of positive test, HbA2 estimation done for confirmation.
- HbA2 estimation is 4-8% and carry out by HPLC.[2]
Differential Diagnosis
Differential Diagnosis:
- Iron deficiency anemia: This is the most common type of anemia and can present with similar symptoms to thalassemia, such as fatigue and pallor. However, iron deficiency anemia is characterized by low levels of iron in the blood, whereas thalassemia is characterized by abnormal hemoglobin production.
- Sideroblastic anemias: These are a group of anemias characterized by the presence of ring sideroblasts in the bone marrow. These anemias can cause similar blood test abnormalities to thalassemia, but they are caused by different underlying mechanisms.
- Hemoglobinopathies: These are a group of inherited disorders that affect the structure or production of hemoglobin. Some hemoglobinopathies, such as sickle cell disease, can cause similar symptoms to thalassemia. However, hemoglobinopathies are characterized by specific abnormalities in the hemoglobin protein, whereas thalassemia is characterized by reduced production of normal hemoglobin.
- Other inherited anemias: There are a number of other inherited anemias that can cause similar symptoms to thalassemia. These include hereditary spherocytosis, hereditary elliptocytosis, and pyruvate kinase deficiency. However, these anemias are caused by different underlying mechanisms than thalassemia.[6]
Complications
Complication:
Possible complications of moderate to severe include i.e.:
- Iron overload
- Infection
- Bone deformities
- Enlarged spleen
- Slowed growth rates-Anemia can both slow a child’s growth and delay puberty.
- Heart problems- Congestive heart failure and abnormal heart rhythms can associate with severe thalassemia.[2]
Investigations
Investigation:
Hematological
- Anemia – Moderate to severe with 10-12 gm/dl. RBCs are microcytic hypochromic. Target cells are present and basophilic stippling is common. Also presence of tear drop, elliptical, fragments in red cells and at times red cell with Howell Jolly body.
Reticulocytotic i.e.
- Leukocytosis with few metamyelocytes also myelocytes
Biochemical
- Reduced serum haptoglobins
- Bilirubin (unconjugated) increased and urine urobilinogen increased
- Iron status –
(a) Serum iron and ferratin markedly increased.
(b) Total iron binding capacity (in other words, TIBC) reduced.
Bone marrow
- Erythroid hyperplasia with reversed M:E ratio
- Normoblastic erythropoiesis
- Ineffective erythropoiesis – Some norm oblasts die in the marrow without maturing into red cells
- Myelopoiesis also Megakaryopoiesis
- Increased bone marrow iron
Other special tests:
- HbF levels are high.
- Hb electrophoresis – Bands of both HbA and HbF in β-thalassemia.
- Global chain synthesis – α-β globin chain synthesis ratio altered (normal l:l) due to lack of synthesis of β chains.
- DNA analysis – useful for predicting disease severity and diagnosis.
- Liver spectrometry for detecting hemosiderosis of liver[2]
Treatment
Treatment:
- Blood transfusions to keep Hb between 9-11 gm%, if infant’s Hb. 6-7 gm% and failure to thrive. Transfusions give every 2-4 weeks.
- Iron chelation – Desferrioxamine (DFX) as S.C. infusion using a syringe driver pump/infuser.
Indications i.e.–
(a) S. ferritin level > 200 mg/L.
(b) Deferiprone orally fairly well tolerate. S. ferritin needs monitoring.
(c) Deferasirox orally, if Deferiprone is not given because of side-effects. Side-effects include arthralgia, anorexia, liver dysfunction.
- Splenectomy – Hypersplenism due to splenomegaly causes neutropenia and increased need for blood transfusion. Splenectomy reduces severity of neutropenia and subsequent infections.
- Bone marrow transplantation – Indication for BMT is in cases where matched siblings are available in a family, if not available, to look for a matched related donor. This is curative for the patient.[2]
Prevention
Prevention:
- Thalassemia trait in parents – If antenatal check up reveals trait (by assessing HbA2 levels (4-8%), then father should assess for thalassemia trait and, if positive then CVS (chronic villous sampling) is necessary.
- Antenatal VS – at 9-10 weeks of gestation analysis of foetal DNA. If fetus is homozygous for thalassemia, termination of pregnancy is advisable.
- Thalassemia screening – All mothers with Hb < 11gm% during their first antenatal check-up should screen for Hb estimation. If MCV < 70 fl, MCH < 23 and RBC count of 7.5 million. If HbA2 estimation 4-8% a diagnosis suggest.[2]
Homeopathic Treatment
Homeopathic Treatment:
Homeopathy treats the person as a whole. It means that homeopathic treatment focuses on the patient as a person, as well as his pathological condition. The homeopathic medicines selected after a full individualizing examination and case-analysis.
which includes
- The medical history of the patient,
- Physical and mental constitution,
- Family history,
- Presenting symptoms,
- Underlying pathology,
- Possible causative factors etc.
A miasmatic tendency (predisposition/susceptibility) also often taken into account for the treatment of chronic conditions.
What Homoeopathic doctors do?
A homeopathy doctor tries to treat more than just the presenting symptoms. The focus is usually on what caused the disease condition? Why ‘this patient’ is sick ‘this way’?.
The disease diagnosis is important but in homeopathy, the cause of disease not just probed to the level of bacteria and viruses. Other factors like mental, emotional and physical stress that could predispose a person to illness also looked for. No a days, even modern medicine also considers a large number of diseases as psychosomatic. The correct homeopathy remedy tries to correct this disease predisposition.
The focus is not on curing the disease but to cure the person who is sick, to restore the health. If a disease pathology not very advanced, homeopathy remedies do give a hope for cure but even in incurable cases, the quality of life can greatly improved with homeopathic medicines.
Homeopathic Medicines for Thalassemia:
The homeopathic remedies (medicines) given below indicate the therapeutic affinity but this is not a complete and definite guide to the homeopathy treatment of this condition. The symptoms listed against each homeopathic remedy may not be directly related to this disease because in homeopathy general symptoms and constitutional indications also taken into account for selecting a remedy.
Medicines:
Alumina
- Generally, Anaemia and chlorosis in young girls at puberty.
- Menses pale also scanty, with abnormal cravings for indigestible things.
- Lastly, Profuse aluminous leucorrhoea.
Argentum nitricum
- Shortness of breath, without either lungs or heart being affected.
- Furthermore, Sallow complexion from defective oxidation of the blood.
- Heart- burn, dyspepsia; irritative flatulent gastralgia; additionally, round ulcer of stomach.
- Menses irregular, either scanty or copious; spinal irritation, albuminuria, tendency to diarrhoea.
- Constant desire especially for candy or sugar.
China
- Complaints specifically from loss of animal fluids, be it blood, semen, diarrhoea, leucorrhoea or over lactation.
- Moreover, Great debility, trembling, aversion to exercise.
- Palpitations with rush of blood to head, additionally redness of face with cold hands.
- Heaviness of head, with loss of sight, fainting also ringing in ears.
- Sleeplessness; In detail, intolerance of fruits.
Ferrum met
- Basically, Pure anemia with appearance of false plethora.
- Face ashy either pale or greenish, becomes bright red in flushes, great paleness of mucous membranes.
- Bellow’s sound of the heart also anaemic murmurs of the arteries and veins.
- Vomiting as soon as food is taken, with relief of gastralgia pains, also prostration with lethargic dullness.
- Animal food not desired, nor is it well borne by the stomach if taken into it.
- Lastly, Anaemia of chlorotic girls and women.
Kali carb
- Frequent chilliness, every time patients goes out of doors.
- Patient becomes chilly from deficiency of red blood-corpuscles in the blood.
- Vertigo when turning head rapidly or from riding in a carriage, with humming in ears.
- Weakness of sight from sexual indulgence.
Natrum Mur
- Blood impoverished; anaemia from loss of fluids; additionally malarious cachexia; emaciation.
- Skin harsh, dry, yellow; great exhaustion from any little exertion of either mind or body.
- Palpitation, with sensation as if a bird’s wing were fluttering in left chest.
- Pressure and distension of stomach; constipation, with contraction of anus; terrible sadness.
Natrum carb
- Pallid anaemia, with great debility, Milky-white skin.
- Vitality below par; emaciation; nervousness also anxiety.
- Aggravation during a thunder storm.
- Playing on either piano or hearing music makes her nervous.
- Inertia in psoric, phlegmatic persons.
Diet & Regimen
Diet & Regimen:
Dietary Recommendations:
- Iron-Restricted Diet: Individuals with thalassemia often experience iron overload due to frequent blood transfusions. Therefore, it is essential to limit iron intake from food sources. This includes avoiding iron-rich foods such as red meat, organ meats, fortified cereals, and certain vegetables like spinach. Consulting a registered dietitian or a healthcare professional specializing in thalassemia is recommended to create a personalized iron-restricted diet plan.
- Focus on Nutrient-Rich Foods: A diet rich in fruits, vegetables, whole grains, and lean protein sources is recommended for individuals with thalassemia. These foods provide essential nutrients and antioxidants that support overall health and well-being.
- Adequate Calcium and Vitamin D Intake: Calcium and vitamin D are vital for bone health, especially in individuals with thalassemia who may be at risk for osteoporosis. Including calcium-rich foods like dairy products, leafy green vegetables, and fortified foods, along with adequate sunlight exposure or vitamin D supplementation, is crucial.
- Hydration: Staying well-hydrated is essential for individuals with thalassemia. Drinking plenty of water throughout the day helps maintain adequate blood volume and prevent complications.
Lifestyle Modifications:
- Regular Exercise: Engaging in regular physical activity helps improve cardiovascular health, maintain a healthy weight, and enhance overall well-being. However, it is essential to consult a healthcare professional to determine the appropriate exercise regimen based on individual health conditions.
- Infection Prevention: Individuals with thalassemia are more susceptible to infections. Practicing good hygiene, such as frequent handwashing and avoiding close contact with sick individuals, is crucial.
- Regular Medical Checkups: Regular follow-up with a healthcare professional specializing in thalassemia is essential to monitor the condition, manage complications, and adjust treatment plans as needed.[4]
Do’s and Don'ts
The Do’s And Don’ts:
Do’s:
- Follow a balanced, low-iron diet. Focus on fruits, vegetables, whole grains, and lean protein. Limit iron-rich foods like red meat, liver, and fortified cereals. Consult a dietitian for a personalized meal plan.
- Stay hydrated. Drink plenty of water throughout the day to maintain adequate blood volume and prevent complications.
- Get regular exercise. Engage in physical activity suitable for your condition to improve cardiovascular health and overall well-being. Consult your doctor before starting any new exercise program.
- Practice good hygiene. Wash your hands frequently and avoid close contact with sick people to prevent infections.
- Get vaccinated. Keep up-to-date on vaccinations, including those for flu, pneumonia, and hepatitis B, as recommended by your doctor.
- Attend regular medical checkups. See your doctor regularly to monitor your condition, manage complications, and adjust treatment plans as needed.
- Take medications as prescribed. If you are prescribed medications for thalassemia or related complications, take them exactly as directed by your doctor.
- Seek emotional support. Connect with other people with thalassemia or join a support group to share experiences and coping strategies.
- Educate yourself about your condition. Learn as much as you can about thalassemia to actively participate in your treatment and make informed decisions.
Don’ts:
- Don’t take iron supplements. Unless specifically prescribed by your doctor, avoid iron supplements and multivitamins containing iron.
- Not smoke or use tobacco products. Smoking can worsen anemia and increase the risk of complications.
- Don’t drink excessive alcohol. Alcohol can interfere with iron absorption and contribute to liver damage.
- Don’t self-medicate. Consult your doctor before taking any over-the-counter medications or supplements, as some may interact with your thalassemia treatment.
- Never ignore signs of infection. Seek medical attention promptly if you develop fever, chills, or other symptoms of infection.
- Don’t hesitate to ask for help. If you are struggling with the physical or emotional challenges of thalassemia, reach out to your healthcare team or a support group for assistance.
Terminology
Terminology:
- Thalassemia: A group of inherited blood disorders characterized by reduced or absent production of hemoglobin, the protein in red blood cells that carries oxygen.
- Hemoglobin: A protein in red blood cells that carries oxygen from the lungs to the body’s tissues and carbon dioxide back to the lungs.
- Anemia: A condition in which the blood has a lower than normal number of red blood cells or a lower than normal amount of hemoglobin. This can lead to fatigue, weakness, and shortness of breath.
- Alpha thalassemia: A type of thalassemia caused by reduced or absent production of the alpha globin chains of hemoglobin.
- Beta thalassemia: A type of thalassemia caused by reduced or absent production of the beta globin chains of hemoglobin.
Other Examination :
- Thalassemia major: A severe form of thalassemia, also known as Cooley’s anemia, that requires lifelong blood transfusions and chelation therapy to manage iron overload.
- Thalassemia intermedia: A moderate form of thalassemia that may require occasional blood transfusions and other treatments.
- Thalassemia minor: A mild form of thalassemia that usually does not require treatment.
- Blood transfusion: A procedure in which blood is given to a person through a vein.
- Chelation therapy: A treatment that removes excess iron from the body.
- Iron overload: A condition in which there is too much iron in the body. This can damage organs such as the heart, liver, and pancreas.
- Splenectomy: Surgical removal of the spleen.
- Bone marrow transplant: A procedure in which healthy bone marrow is transplanted from a donor to a recipient.
- Gene therapy: A treatment that involves introducing healthy genes into a person’s cells to replace faulty genes.
- Carrier: A person who has one copy of a thalassemia gene but does not have the condition. Carriers can pass the gene on to their children.
- Genetic counseling: A process in which a healthcare professional provides information and support to people who have, or are at risk of having, a genetic condition.
Homoeopathic Terminology:
- Anemia: A condition where the blood lacks enough healthy red blood cells or hemoglobin to carry adequate oxygen to the body’s tissues. It is a common symptom of thalassemia.
- Constitutional Remedy: A homeopathic remedy chosen based on the patient’s overall physical, mental, and emotional characteristics, aiming to treat the underlying cause of the disease rather than just the symptoms.
- Genetic Predisposition: An increased likelihood of developing a particular disease based on inherited genes. Thalassemia is a genetic blood disorder.
- Hemoglobin: The iron-containing protein in red blood cells that carries oxygen from the lungs to the body’s tissues. Thalassemia results in decreased or abnormal hemoglobin production.
- Iron Overload: A condition where excess iron builds up in the body. It is a common complication of thalassemia due to frequent blood transfusions.
Other Terminology:
- Miasm: In homeopathy, a miasm is a theoretical underlying predisposition to chronic disease. Homeopaths believe that understanding a patient’s miasm can help guide treatment selection.
- Potency: The strength or dilution of a homeopathic remedy. Homeopathic remedies are prepared through a process of serial dilution and succussion, and higher potencies are believed to have deeper acting effects.
- Repertory: A reference book used by homeopaths to find remedies based on specific symptoms.
- Similia Similibus Curentur: The principle of "like cures like" that is the foundation of homeopathy. It suggests that a substance that can cause symptoms in a healthy person can also cure similar symptoms in a sick person.
- Vital Force: In homeopathy, the vital force is the energy or life force that animates the body and maintains health. Homeopaths believe that disease arises from disturbances in the vital force, and that homeopathic remedies act by stimulating the vital force to restore health.
References
References:
- Medicine Golwala
- https://www.mayoclinic.org/diseasesconditions/thalassemia/symptoms-cause
- "Prevalence and Management of β-Thalassemia in India", Tandfonline, 2021”
- Hematology: Basic Principles and Practice (7th Edition)
- Hoffbrand, A. V., Moss, P. A. H., & Pettit, J. E. (2006). Essential Haematology (5th ed.). Blackwell Publishing.
- Wintrobe’s Clinical Hematology,14th Edition
Also Search As
Also Searched As:
Search engines:
- Use specific keywords: Start with basic searches using terms like "homeopathy thalassemia" or "homeopathic treatment thalassemia." Then, refine your search by adding specific keywords related to the aspect of thalassemia or homeopathy you’re interested in, such as "case studies," "clinical trials," "remedies," or "research."
- Utilize advanced search operators: To narrow down your search further, use advanced search operators like quotation marks for exact phrases (e.g., "thalassemia major"), the minus sign to exclude specific terms (e.g., homeopathy -conventional medicine), or the site: operator to search within a specific website (e.g., site:.edu homeopathy thalassemia).1. Refine Google searches – Google Help support.google.com
Specialized databases and websites:
- Homeopathic journals and publications: Search within online databases of reputable homeopathic journals and publications like "Homeopathy" or "The American Journal of Homeopathic Medicine." 1. Research articles – Faculty of Homeopathy www.facultyofhomeopathy.org
- Homeopathic organizations and associations: Explore the websites of homeopathic organizations and associations, both national and international, as they often provide resources and articles on various health conditions, including thalassemia.
- ResearchGate and PubMed: These platforms host a vast collection of scientific publications and research papers, including those on homeopathy and thalassemia. 1. Over 150 million publication pages and growing: how ResearchGate is bringing more research to the platformwww.researchgate.net
General Searches
- "Thalassemia" – This simple search will provide a broad overview of thalassemia, including basic information, symptoms, causes, and treatments.
- "Thalassemia overview" or "Thalassemia introduction" – These searches will likely return introductory articles or educational materials on thalassemia.
Specific Types of Thalassemia
- "Alpha thalassemia" or "Beta thalassemia" – These searches will focus on specific types of thalassemia, providing information on their unique characteristics, symptoms, and treatment options.
- "Thalassemia major" or "Thalassemia intermedia" – These searches will target articles on the severity of thalassemia, detailing the associated symptoms, complications, and treatment needs.
Specific Aspects of Thalassemia
- "Thalassemia symptoms" or "Thalassemia complications" – These searches will focus on the clinical manifestations of thalassemia, highlighting the signs and symptoms to look out for.
- "Thalassemia treatment" or "Thalassemia management" – These searches will focus on the various therapeutic options available for thalassemia, including blood transfusions, iron chelation therapy, and potential cures like bone marrow transplants.
- "Thalassemia diet" or "Thalassemia nutrition" – These searches will provide information on dietary recommendations for individuals with thalassemia, including iron-restricted diets and nutrient-rich food choices.
- "Thalassemia genetics" or "Thalassemia inheritance" – These searches will explore the genetic basis of thalassemia, explaining how the condition is passed down from parents to children.
- "Thalassemia research" or "Thalassemia clinical trials" – These searches will uncover the latest advancements in thalassemia research and ongoing clinical trials, providing insights into potential new treatments and therapies.
Frequently Asked Questions (FAQ)
What is Thalassemia?
They are genetic disorders of Hb synthesis in which there is reduced production of one or more chains of Hb.
Can thalassemia be cured?
- Currently, the only potential cure for thalassemia is a bone marrow transplant. However, this procedure is complex and carries risks.
What causes Thalassemia?
It is caused by mutations in the DNA of cells that make hemoglobin — the substance in red blood cells that carries oxygen throughout your body
What are the types of Thalassemia?
α
β
Can homeopathy cure thalassemia?
- While homeopathy cannot cure thalassemia, it may offer supportive treatment to manage symptoms, improve quality of life, and potentially reduce the frequency of blood transfusions.
What are the symptoms of Thalassemia?
- Anemic
- Increasing pallor
- Splenomegaly
- Frontal bossing
- Mild hemolytic jaundice
- Bone changes- ‘Hair on end’ appearance
- Increased susceptibility to infections
- Hepatomegaly
- Cardiac involvement– Myocardial hemosiderosis
How does homeopathy work in thalassemia?
- Homeopathy aims to stimulate the body’s natural healing abilities. A homeopath will select a remedy based on the individual’s unique symptoms and constitution.
How long does it take to see results with homeopathy for thalassemia?
- The response to homeopathic treatment can vary depending on the individual and the severity of the condition. Some people may experience improvement in symptoms within a few weeks, while others may take longer.
Homeopathic Medicines used by Homeopathic Doctors in treatment of Thalassemia?
- Alumina
- Argentum nitricum
- China
- Ferrum met
- Kali carb
- Natrum Mur
- Natrum carb